
OGDH





Alpha-ketoglutarate dehydrogenase also known as 2-oxoglutarate dehydrogenase E1 component, mitochondrial is an enzyme that in humans is encoded by the OGDH gene.[5][6][7]
Structure[edit]
Gene[edit]
The OGDH gene is located on the 7th chromosome, with the specific location being 7p14-p13. There are 26 exons located within the gene.[7]
Protein[edit]
This gene encodes a subunit that catalyzes the oxidative decarboxylation of alpha-ketoglutarate to Succinyl-CoA at its active site in the fourth step of the citric acid cycle by acting as a base to facilitate the decarboxylation. The main residues responsible for the catalysis are thought to be His 260, Phe 227, Gln685, His 729, Ser302, and His 298.[8]
Function[edit]
This gene encodes one subunit of the 2-oxoglutarate dehydrogenase complex. This complex catalyzes the overall conversion of 2-oxoglutarate (alpha-ketoglutarate) to succinyl-CoA and CO2 during the citric acid cycle. The protein is located in the mitochondrial matrix and uses thiamine pyrophosphate as a cofactor.[7] The overall complex furthers catalysis by keeping the necessary substrates for the reaction close within the enzyme, thus creating a situation in which it is more likely that the substrate will be in the favorable conformation and orientation. This enzyme is also part of a larger multienzyme complex that channels the intermediates in the catalysis between subunits of the complex thus minimizing unwanted side reactions. Not only do the subunits ferry products back and forth, but each of the subunits in the E1o homodimer are connected via a cavity lined with acidic residues, thus increasing the dimer's ability to act as a base. The orientation of the cavity allows for direct transfer of the intermediate to the E2o subunit.[9]
Mechanism[edit]
The protein encoded by OGDH is thought to have a single active site. The enzyme also requires two cofactors in order for it to function properly, Thiamine diphosphate and a divalent magnesium ion. The specific mechanism of the subunit is currently unknown; however, there are several theories as to how it functions, among them is the Hexa Uni Ping Pong theory.[10] Even though the mechanism isn't fully known the kinetic data have been calculated and are as follows: the Km is 0.14 ± 0.04 mM, and the Vmax is 9 ± 3 μmol/(min*mg).[11]
Regulation[edit]
This subunit, known as E1o, catalyzes a rate-limiting step in the citric acid cycle and lies far from equilibrium; the total change in Gibbs free energy is ΔG = −33 kJ/mol. The significant energy change makes it a crucial point of regulation not only for the citric acid cycle, but also for the entire cellular respiration pathway. As such, E1o is inhibited by both NADH and Succinyl-CoA via non competitive feedback inhibition.[8]
Clinical significance[edit]
A congenital deficiency in 2-oxoglutarate dehydrogenase activity is believed to lead to hypotonia, metabolic acidosis, and hyperlactatemia. It is characterized by the buildup of a chemical called lactic acid in the body and a variety of neurological problems. Signs and symptoms of this condition usually first appear shortly after birth, and they can vary widely among affected individuals. The most common feature is a potentially life-threatening buildup of lactic acid (lactic acidosis), which can cause nausea, vomiting, severe breathing problems, and an abnormal heartbeat. People with pyruvate dehydrogenase deficiency usually have neurological problems as well. Most have delayed development of mental abilities and motor skills such as sitting and walking. Other neurological problems can include intellectual disability, seizures, weak muscle tone (hypotonia), poor coordination, and difficulty walking. Some affected individuals have abnormal brain structures, such as underdevelopment of the tissue connecting the left and right halves of the brain (corpus callosum), wasting away (atrophy) of the exterior part of the brain known as the cerebral cortex, or patches of damaged tissue (lesions) on some parts of the brain. Because of the severe health effects, many individuals with pyruvate dehydrogenase deficiency do not survive past childhood, although some may live into adolescence or adulthood.[7]
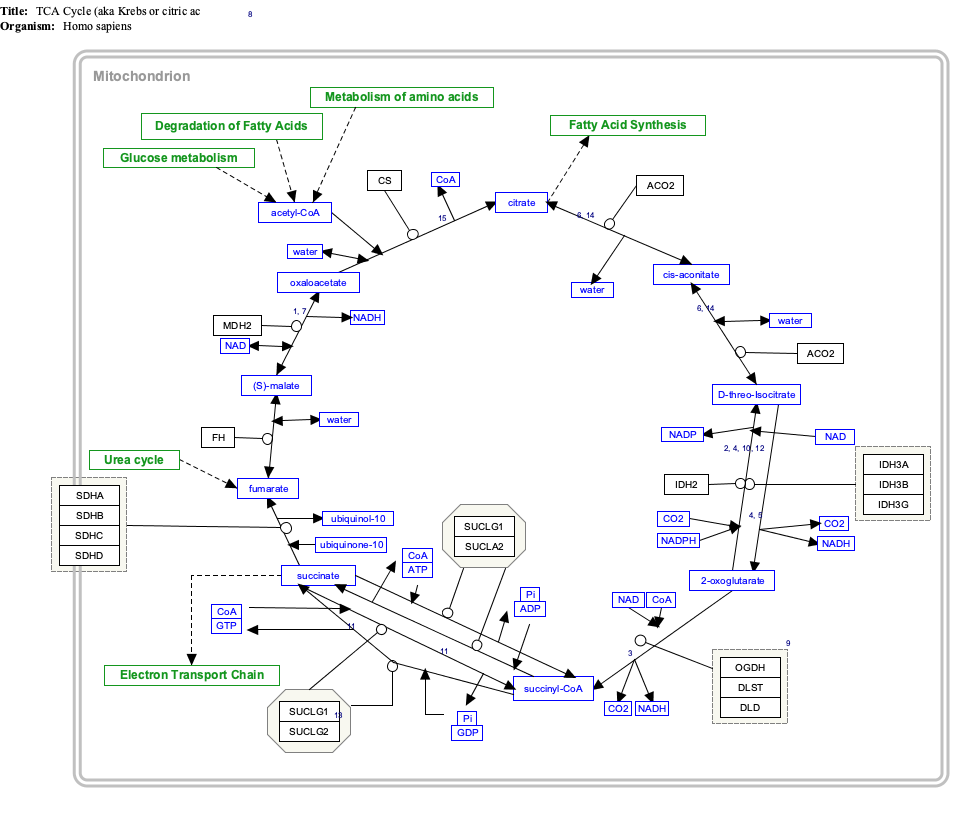
Interactive pathway map[edit]
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
{kind=link}
- ^ The interactive pathway map can be edited at WikiPathways: "TCACycle_WP78".
References[edit]
- ^ a b c GRCh38: Ensembl release 89: ENSG00000105953 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000020456 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Koike K, Urata Y, Goto S (Mar 1992). "Cloning and nucleotide sequence of the cDNA encoding human 2-oxoglutarate dehydrogenase (lipoamide)". Proceedings of the National Academy of Sciences of the United States of America. 89 (5): 1963–7. Bibcode:1992PNAS...89.1963K. doi:10.1073/pnas.89.5.1963. PMC 48574. PMID 1542694.
- ^ Szabo P, Cai X, Ali G, Blass JP (Mar 1994). "Localization of the gene (OGDH) coding for the E1k component of the alpha-ketoglutarate dehydrogenase complex to chromosome 7p13-p11.2". Genomics. 20 (2): 324–6. doi:10.1006/geno.1994.1178. PMID 8020988.
- ^ a b c d "Entrez Gene: oxoglutarate (alpha-ketoglutarate) dehydrogenase (lipoamide)".
- ^ a b Frank RA, Price AJ, Northrop FD, Perham RN, Luisi BF (May 2007). "Crystal structure of the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex". Journal of Molecular Biology. 368 (3): 639–51. doi:10.1016/j.jmb.2007.01.080. PMC 7611002. PMID 17367808.
- ^ Voet DJ, Voet JG, Pratt CW (2010). "Chapter 18, Mitochondrial ATP synthesis". Principles of Biochemistry (4th ed.). Wiley. p. 669. ISBN 978-0-470-23396-2.
- ^ McMinn CL, Ottaway JH (Mar 1977). "Studies on the mechanism and kinetics of the 2-oxoglutarate dehydrogenase system from pig heart". The Biochemical Journal. 161 (3): 569–81. doi:10.1042/bj1610569. PMC 1164543. PMID 192200.
- ^ Leung PS, Rossaro L, Davis PA, Park O, Tanaka A, Kikuchi K, Miyakawa H, Norman GL, Lee W, Gershwin ME (Nov 2007). "Antimitochondrial antibodies in acute liver failure: implications for primary biliary cirrhosis". Hepatology. 46 (5): 1436–42. doi:10.1002/hep.21828. PMC 3731127. PMID 17657817.
Further reading[edit]
- Shi Q, Chen HL, Xu H, Gibson GE (Mar 2005). "Reduction in the E2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity". The Journal of Biological Chemistry. 280 (12): 10888–96. doi:10.1074/jbc.M409064200. PMID 15649899.
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M (Oct 2005). "Towards a proteome-scale map of the human protein-protein interaction network". Nature. 437 (7062): 1173–8. Bibcode:2005Natur.437.1173R. doi:10.1038/nature04209. PMID 16189514. S2CID 4427026.
- Reed LJ, Hackert ML (Jun 1990). "Structure-function relationships in dihydrolipoamide acyltransferases". The Journal of Biological Chemistry. 265 (16): 8971–4. doi:10.1016/S0021-9258(19)38795-2. PMID 2188967.
- Sanger Centre, The; Washington University Genome Sequencing Cente, The (Nov 1998). "Toward a complete human genome sequence". Genome Research. 8 (11): 1097–108. doi:10.1101/gr.8.11.1097. PMID 9847074.
- Bonaldo MF, Lennon G, Soares MB (Sep 1996). "Normalization and subtraction: two approaches to facilitate gene discovery". Genome Research. 6 (9): 791–806. doi:10.1101/gr.6.9.791. PMID 8889548.
- Koike K (Jul 1995). "The gene encoding human 2-oxoglutarate dehydrogenase: structural organization and mapping to chromosome 7p13-p14". Gene. 159 (2): 261–6. doi:10.1016/0378-1119(95)00086-L. PMID 7622061.
- Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, Saito K, Isono Y, Irie R, Kushida N, Yoneyama T, Otsuka R, Kanda K, Yokoi T, Kondo H, Wagatsuma M, Murakawa K, Ishida S, Ishibashi T, Takahashi-Fujii A, Tanase T, Nagai K, Kikuchi H, Nakai K, Isogai T, Sugano S (Jan 2006). "Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes". Genome Research. 16 (1): 55–65. doi:10.1101/gr.4039406. PMC 1356129. PMID 16344560.
- McCartney RG, Rice JE, Sanderson SJ, Bunik V, Lindsay H, Lindsay JG (Sep 1998). "Subunit interactions in the mammalian alpha-ketoglutarate dehydrogenase complex. Evidence for direct association of the alpha-ketoglutarate dehydrogenase and dihydrolipoamide dehydrogenase components". The Journal of Biological Chemistry. 273 (37): 24158–64. doi:10.1074/jbc.273.37.24158. PMID 9727038.
- van Bever Y, Balemans W, Duval EL, Jespers A, Eyskens F, van Hul W, Courtens W (Apr 2007). "Exclusion of OGDH and BMP4 as candidate genes in two siblings with autosomal recessive DOOR syndrome". American Journal of Medical Genetics Part A. 143A (7): 763–7. doi:10.1002/ajmg.a.31641. PMID 17343268. S2CID 11529600.
- Habelhah H, Laine A, Erdjument-Bromage H, Tempst P, Gershwin ME, Bowtell DD, Ronai Z (Dec 2004). "Regulation of 2-oxoglutarate (alpha-ketoglutarate) dehydrogenase stability by the RING finger ubiquitin ligase Siah". The Journal of Biological Chemistry. 279 (51): 53782–8. doi:10.1074/jbc.M410315200. PMID 15466852.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|